I feel that I’m somehow misunderstanding the proximity filter.
(Running a fully updated version of nextstrain software and using a github directory pull from this week)
What I’m trying to do is hand Nextstrain a small number of pre-defined sequences, and have it spit out a tree of genetically-similar sequences. For this trial-run, I took the GISAID database, filtered it to USA-only samples, and marked three samples (which all came from the same exposure event and are all delta variant sequences from mid-June 2021) with a new column in the metadata. Then I asked nextstrain to construct a build that had 100 sequences in these samples’ proximity:
subsampling:
target:
testfocus:
exclude: "--exclude-where 'testcolumn!=focus'"
testproximity:
max_sequences: 100
priorities:
type: "proximity"
focus: "testfocus"
First I had a problem where it was ignoring Wuhan/Hu-1, but not Wuhan/WH01, both of which were being included in the testfocus subsampling. So I manually removed WH01 from my metadata to prevent this issue. The build ran without complaint.
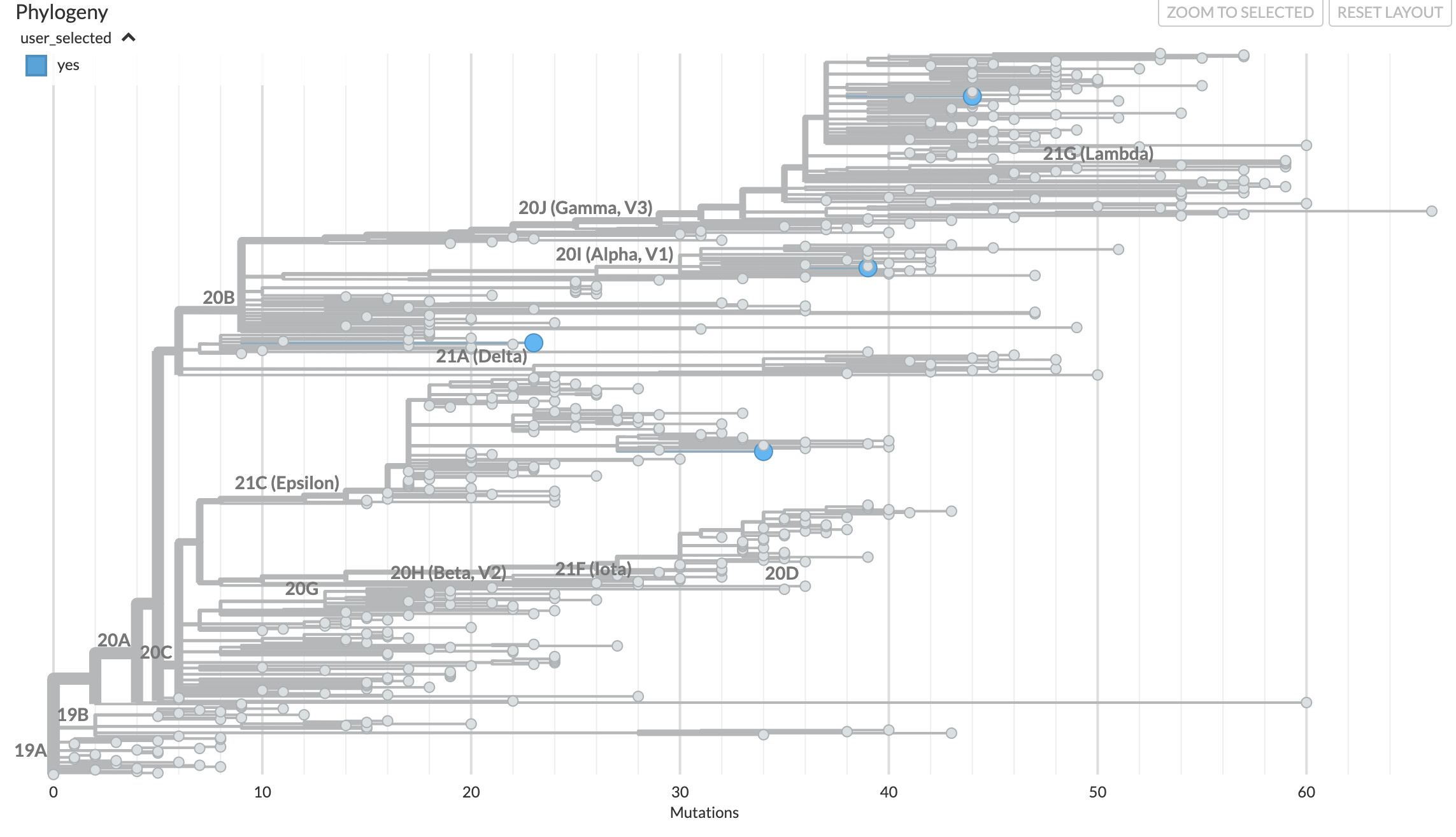
However, I would presume that with >15000 delta variants in the dataset, and my requesting the 100 closest genetic matches, all 100 would naturally be delta variants. What I get instead is mostly 20B and 20C, with only a single delta mixed in:

What am I doing wrong? When I look at proximity_testfocus.tsv, I see a numerical score for each sequence. The ones with the lowest ‘distances’ are the sequences I probably want, right? There are fifty samples with a distance of 0, 1, or 2.
When I look at priority_testfocus.tsv, and sort by the second column, I get priority scores between -1.01 and -6174.04. Here is where I become more unsure, because when I search in this list for the low-distance numbers mentioned above, here’s what I get:
-6170.12
-6171.09
It seems like better matches have more negative values?
When I open up sample-testproximity.txt to see what samples were picked by the algorithm, here are the priority scores of the first five on the list:
-53.15
-53.12
-52.39
-52.22
-47.62
So I am just thoroughly confused about what the algorithm is doing and what I could be doing wrong.
Thank you for your time.